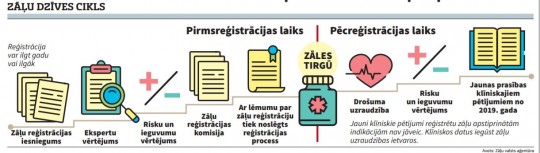
Zāļu dzīve sākas krietni pirms reģistrācijas un turpinās vēl ilgi pacientu zāļu skapīšos, norāda Zāļu valsts aģentūra.
Varētu šķist: ja jau zāles ir nonākušas līdz aptiekas plauktam, tad to izpēte ir pabeigta, tomēr tā nav. Arī pēc medikamentu nonākšanas tirgū joprojām tiek apkopota informācija par zāļu drošuma jautājumiem - blakusparādībām, klīniskiem novērojumiem u.c. Ja pēcreģistrācijas laikā tiek fiksēti dati par jauniem zāļu riskiem, valsts iestādes reaģē nekavējoties.
Ekrānšāviņš no avīzes
Pēdējo gadu zināmākie precedenti ir Remantadīna klasificēšana par recepšu zālēm (līdz 2016. gadam tās bija bezrecepšu zāles), kā arī zāļu Bioparox atsaukšana no tirgus visā Eiropas Savienībā, jo eksperti konstatēja, ka šā medikamenta radītie riski neatsver ieguvumus. Tādu piemēru ir daudz.
Procesa sākumā ir zāļu reģistrācijas apliecības īpašnieka iesniegums un dokumenti par zāļu drošumu, efektivitāti un kvalitāti. Pēc tam seko primārā izvērtēšana - kvalitātes, drošuma un efektivitātes ekspertīze. Šajā posmā zāļu reģistrācijas eksperti vairākkārt aicina dokumentu iesniedzēju atbildēt uz dažādiem jautājumiem, lai zāļu reģistrēšana pilnībā atbilstu visām spēkā esošajām likumu normām. Kad atbildes ir saņemtas, seko ekspertu sākotnējais novērtējums saņemtajai informācijai. Ja šī informācija nav pietiekama, reģistrācijas apliecības īpašniekam tiek atkārtoti lūgts dokumentus pilnveidot. Tā var notikt vairākas reizes, līdz visi iebildumi ir atspēkoti un ZVA ekspertiem nav šaubu par zāļu atbilstību reģistrācijas kritērijiem. Vidēji šis process noris pusgadu un ilgāk. Zāļu valsts aģentūra uzsver, ka riska un ieguvuma līdzsvars tiek noteikts atbilstoši gala dokumentiem, nevis sākotnēji iesniegtajiem materiāliem vai konstatējumiem procesa laikā.
Kvalitātes, drošuma un efektivitātes ekspertīze attīstās līdz ar tehnisko progresu, līdzīgi notiek arī strauja attīstība ārstniecībā un citās jomās. Atbilstoši šai attīstībai tiek pilnveidots arī juridiskais regulējums, sagatavotas zāļu novērtēšanas vadlīnijas, kuras pirms 15 gadiem nebija pieejamas. Taču turpmākās izmaiņas reģistrācijā tiek balstītas uz jauniegūtu informāciju zāļu dzīves cikla laikā. Mainoties videi, cilvēka organisma reakcijām, iegūstot jaunus datus par zālēm, valsts iestādēm, ārstiem un pacientiem līdzdarbojoties, būtiski ir fiksēt signālu par jebkādu neatbilstību. Atbilstoši iegūtajai informācijai ZVA var lemt par papildu darbībām, kas saistītas ar zāļu drošumu, vai jauniem pienākumiem reģistrācijas apliecības īpašniekiem.
Ja notiek izmaiņas prasībās un izpratnē par klīniskajiem pētījumiem, netiek atcelti visi pētījumi, kas veikti pirms prasību izmaiņām. Piemēram, 2019. gadā stāsies spēkā jauni normatīvie akti klīnisko pētījumu jomā, taču tas nenozīmē, ka visām reģistrētajām zālēm būs jāveic jauni pētījumi. Jaunā regula attieksies tikai uz jaunajiem pētījumiem. Savukārt pēc zāļu reģistrācijas to drošums un efektivitāte tiek nodrošināta ar labas ražošanas prakses (GMP - good manufacturing practice - angļu val.) inspekcijām, drošuma uzraudzību, piegādes ķēžu licencēm.